
The Problem with Docking Scores in Drug Discovery
Everyone in drug discovery talks about docking scores. Investors ask about them. Pitch decks are built around them. LinkedIn posts celebrate them.
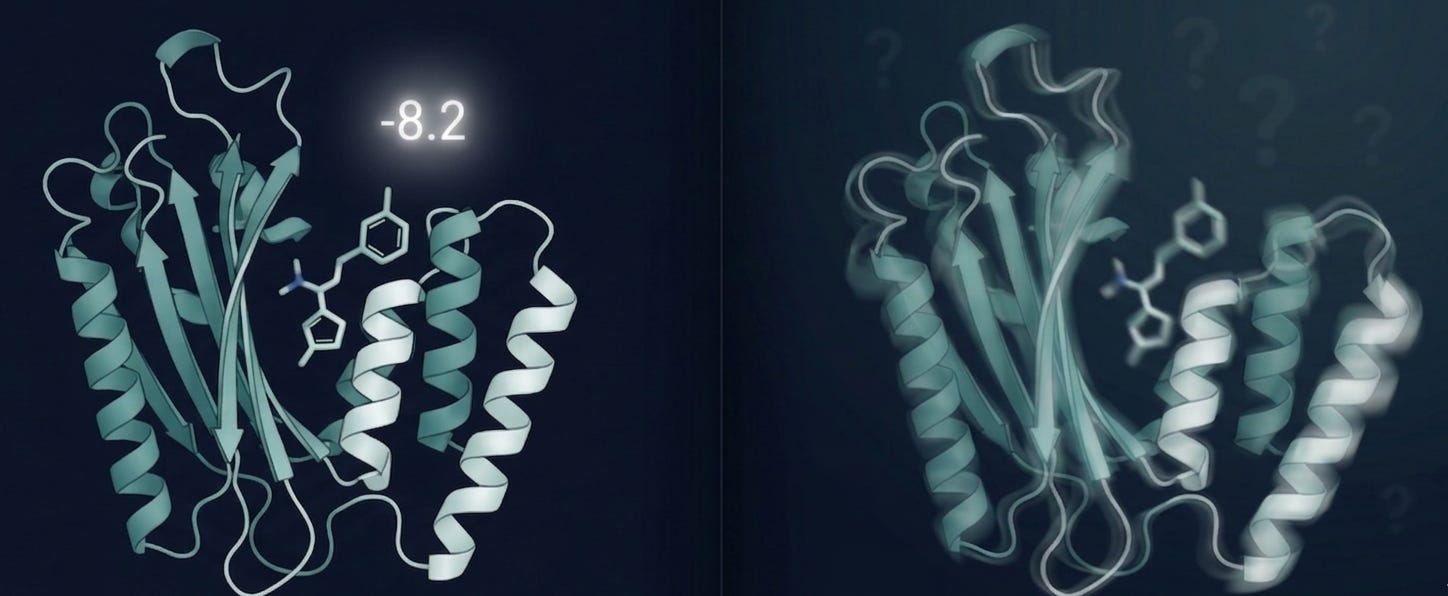
There’s just one problem: a docking score is not a result. It’s a hypothesis.
Let’s talk about what’s actually happening under the hood.
Picture this: you take a protein structure, usually a crystal structure pulled from the PDB, frozen in a single conformation, with missing flexible loops and resolution gaps that leave genuine ambiguity about where atoms sit. You take a small molecule. You run AutoDock Vina, GNINA, GOLD or GLIDE. A number comes out.
That number tells you how well the algorithm thinks the molecule fits the binding pocket given a rigid receptor, implicit solvent, and a scoring function trained on datasets that may or may not look anything like your target class [1,2].
What it doesn’t tell you: whether the molecule will actually bind in a cell. Whether it survives metabolism long enough to get there. Whether your protein even resembles that crystal structure once it’s inside a living system.
Here’s the thing, the field knows this. It just doesn’t say it out loud.
A critical analysis of over 400 published virtual screening studies found that hit rates vary wildly, and that computational filters, binding assays, and secondary validation are routinely skipped before compounds get advanced [3]. In practice, even a well-designed screen might yield around 3 confirmed hits per 100 compounds experimentally tested [4].This isn’t a software problem. It’s a physics problem. Conventional scoring functions struggle to capture long-range electrostatics, desolvation upon binding, and entropic contributions to the precise physical forces that govern whether a molecule actually binds under biological conditions [5]. And most docking algorithms still treat the receptor as a rigid body, even though proteins move constantly between conformational states and ligand binding almost always reshapes the pocket to some degree [6].
The problem isn’t docking. Docking is a powerful, legitimate tool when it’s used as one.
The problem is what happens when a score becomes the story.
So what does rigorous docking actually look like?
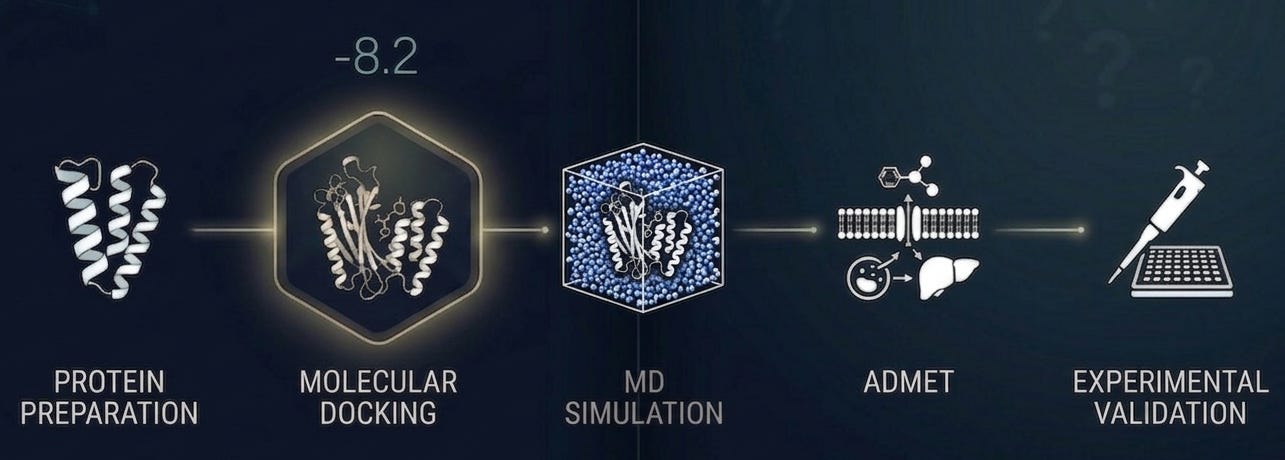
It looks like one step in a workflow, not the whole workflow. It starts with a carefully prepared protein structure: protonation states assigned, missing residues modeled, binding pocket validated before a single compound is screened [5]. It uses multiple scoring functions with consensus scoring to reduce false positives. It accounts for receptor flexibility through ensemble docking or induced-fit protocols where it matters most.
And critically: every top-ranked compound is treated as a hypothesis to interrogate, not a discovery to announce.
The molecules worth pursuing emerge from integrating docking results with ADMET predictions, molecular dynamics stability checks which overcome the rigid sampling limitations inherent in static docking and, wherever possible, experimental data from analogous systems [7].
That pipeline is slower. It produces fewer headlines. But it produces fewer expensive failures two years and fifty million dollars down the road.
Why does this matter right now?
Because as AI-powered docking tools multiply, the temptation to compress this workflow grows louder. Models are faster. Interfaces are cleaner. It has never been easier to generate a ranked list of compounds and call it a drug discovery campaign.
The teams that actually move molecules into the clinic will be the ones who never forgot what a docking score really is and what it isn’t.
At Pauling.AI, that distinction is built into everything we do. Not because rigor is fashionable, but because it’s the only thing that holds up when the biology talks back.
References
Gu, J. et al. (2021). Molecular docking-based computational platform for high-throughput virtual screening. SpringerPlus / PMC. https://pmc.ncbi.nlm.nih.gov/articles/PMC8754542/
Pinzi, L. & Rastelli, G. (2019). Molecular Docking: Shifting Paradigms in Drug Discovery. International Journal of Molecular Sciences, 20(18), 4331. https://pmc.ncbi.nlm.nih.gov/articles/PMC3151162/
Hevener, K.E. et al. (2012). Hit Identification and Optimization in Virtual Screening: Practical Recommendations Based upon a Critical Literature Analysis. Journal of Medicinal Chemistry, 56(3), 927–944. https://pmc.ncbi.nlm.nih.gov/articles/PMC3772997/
Cambridge MedChem Consulting. Virtual Screening Hit Rate Analysis. https://www.cambridgemedchemconsulting.com/resources/hit_identification/virtual_screening_selection.html
Ramírez, D. & Caballero, J. (2020). Practical Considerations in Virtual Screening and Molecular Docking. Frontiers in Chemistry / PMC. https://pmc.ncbi.nlm.nih.gov/articles/PMC7173576/
Totrov, M. & Abagyan, R. (2008). Flexible ligand docking to multiple receptor conformations: a practical alternative. Current Opinion in Structural Biology, 18, 178–184. Cited in: Challenges in Docking: Mini Review. JSciMedCentral. https://www.jscimedcentral.com/jounal-article-info/JSM-Chemistry/Challenges-in-Docking:-Mini-Review-8879
Tao, X. et al. (2022). Integration of Molecular Docking Analysis and Molecular Dynamics Simulations for Studying Food Proteins and Bioactive Peptides. Journal of Agricultural and Food Chemistry. https://pubs.acs.org/doi/10.1021/acs.jafc.1c06110