
The End of Manual MD Simulations: How AI Agents Are Automating GROMACS Workflows
By the Pauling.AI Team.
Automated MD doesn’t change what’s possible in drug discovery. It changes what’s practical.
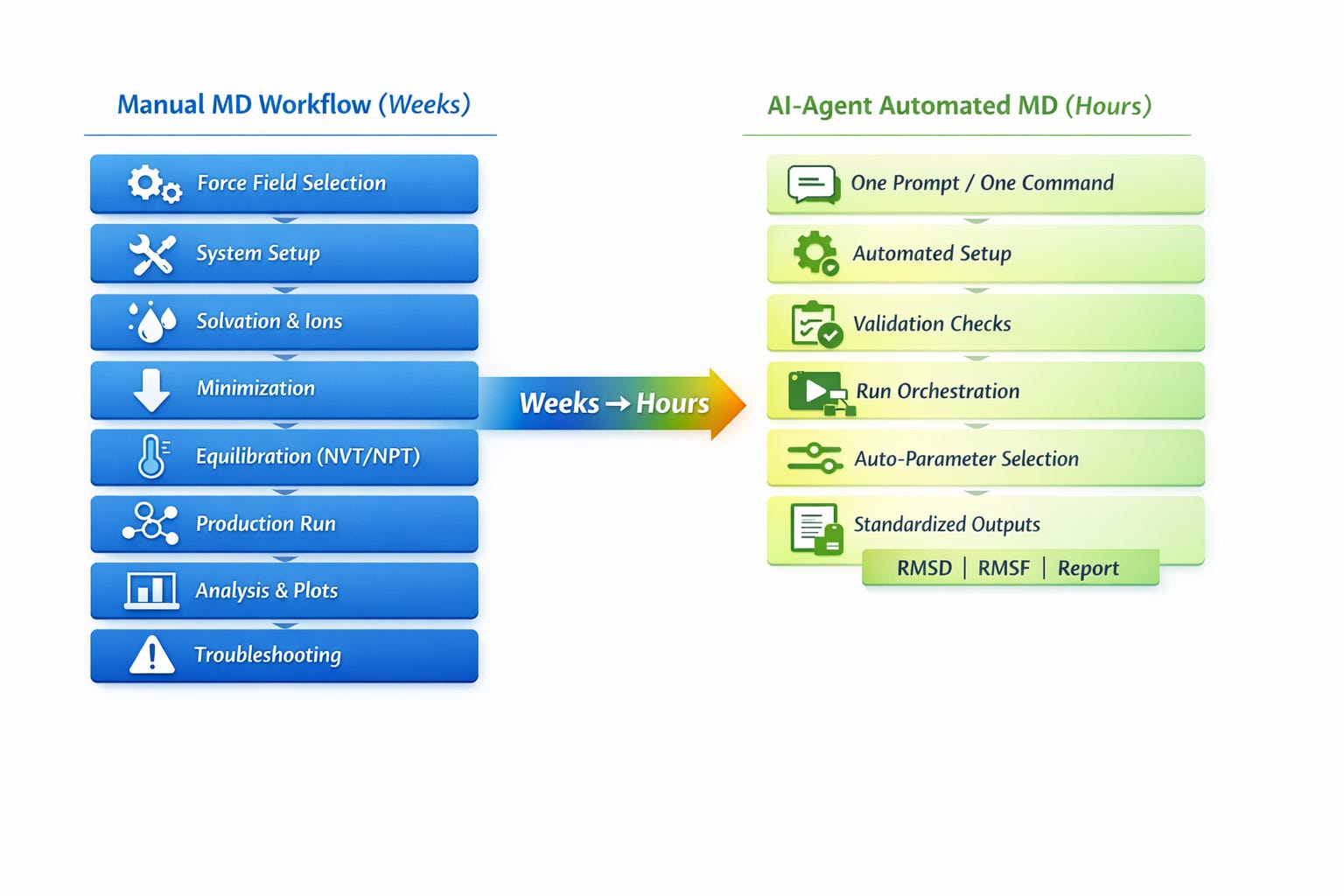
Setting up a molecular dynamics campaign has traditionally meant weeks of manual work before a single production run begins: force field selection, solvation box configuration, equilibration tuning, custom analysis scripting multiplied across every complex in the library. For most computational teams, that overhead isn’t the exception. It’s the baseline.
Automated MD pipelines collapse that timeline. A full campaign complex triage, AMBER and CHARMM force field assignment, TIP3P solvation at 0.15M NaCl, 50,000-step minimization, NVT/NPT equilibration, and 100 ns production runs across 100 complexes in parallel, with binding free energy analysis now runs in roughly 10 compute hours with zero manual intervention.
The science hasn’t changed. The infrastructure has. And the gap between teams that have made that transition and those still running manual workflows widens every quarter.
The Manual Workflow Bottleneck
Running molecular dynamics in GROMACS requires navigating 200+ interdependent parameters across multiple file formats.
Computational chemists face three compounding bottlenecks:
Parameter selection (force fields, water models, thermostat coupling).
Performance tuning (PME ranks, GPU allocation, domain decomposition)
Post-simulation analysis (custom scripting for RMSD, RMSF, binding energies like dG by MMPBSA or FEP.
Studies show manual workflows take “excessive time and effort” [1], and 60-70% of setup goes to chores, not discovery.
AI Agents Enter the Scene
Large language models combined with domain-specific automation offer the solution. A February 2025 study by Lin et al. demonstrates LLM-based systems can automate molecular dynamics workflows through natural language commands [3].
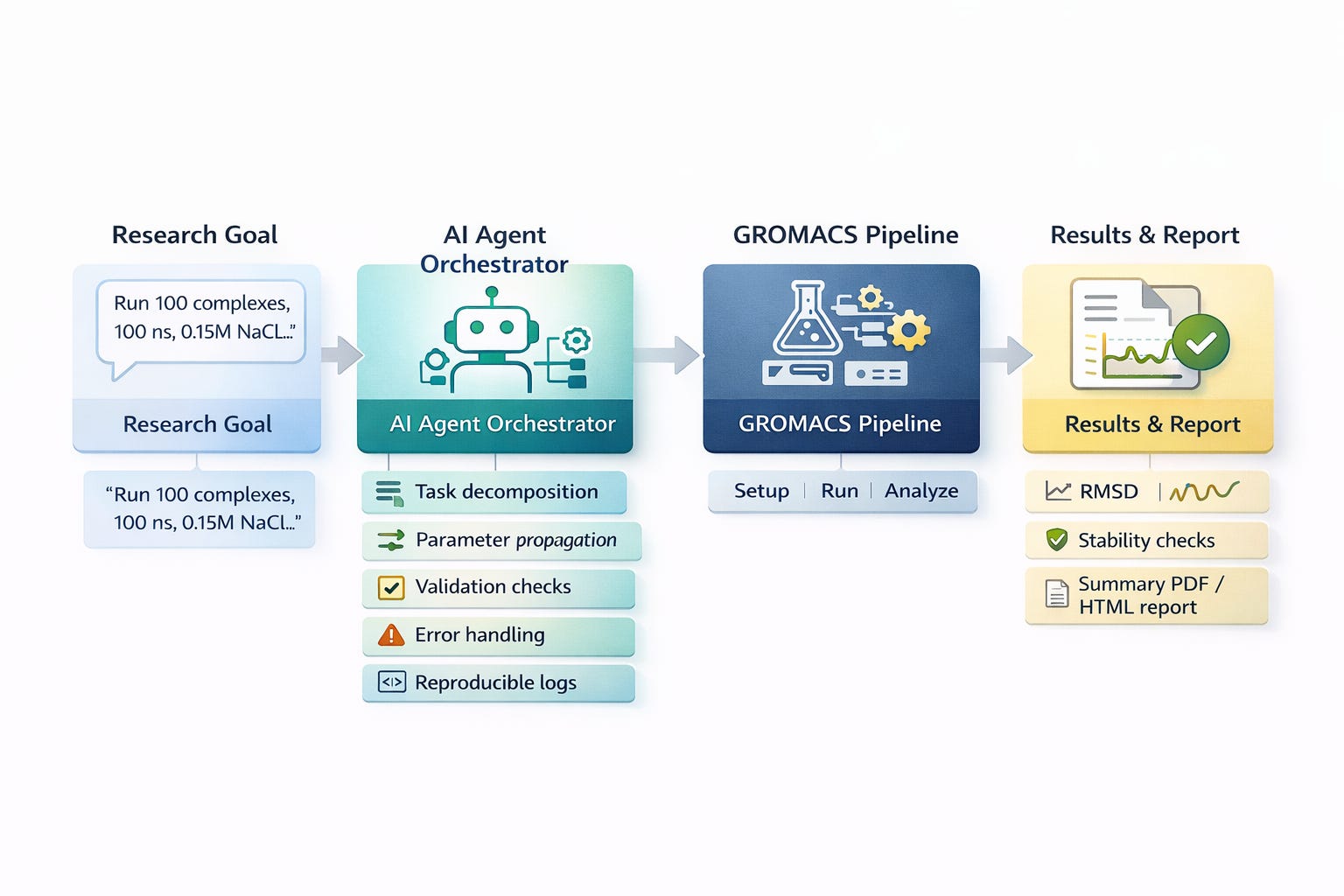
Instead of manually editing configuration files, researchers describe scientific objectives and autonomous agents handle file preparation, parameter selection, simulation execution, and analysis.
Independent research supports this direction: the MDCrow study tested multiple LLM architectures (GPT-4, Claude, Llama), demonstrating that autonomous orchestration of complex GROMACS pipelines is achievable [3].”
Practical implementations are operational. Platforms like Autochem are developing command-based systems that decompose the GROMACS pipeline: complex selection from docked libraries, system preparation with force fields (AMBER, CHARMM, OPLS) and water models (TIP3P, TIP4P, SPC/E), automated solvation, ionization, energy minimization, NVT/NPT equilibration, production runs, and comprehensive analysis.
Dozens of manual gmx commands (pdb2gmx, editconf, solvate, grompp, mdrun) now execute through workflows with intelligent parameter propagation.
The automation spectrum ranges from specialized analysis tools (2024 J. Biomol. Struct. Dyn. [1]) generating reports via single commands to end-to-end platforms (2023 Comput. Struct. Biotechnol. J. [4]) integrating with GROMACS modules. The shift: treating MD workflows as programmable sequences AI executes autonomously.
Intelligent Parameter Selection
AI tackles GROMACS’s most technical challenge: parameter optimization. Traditional “one-factor-at-a-time” tuning fails to capture complex interactions between PME ranks, GPU allocation, and domain decomposition [2]. Research by Kutzner et al. (2019) and Abraham et al. (2025) uses systematic algorithms identifying ideal parameter combinations across hardware from workstations to 65,000-core supercomputers [2][5]. These systems automatically balance PP/PME workloads, configure CPU affinity, and optimize bonded interaction offloading.
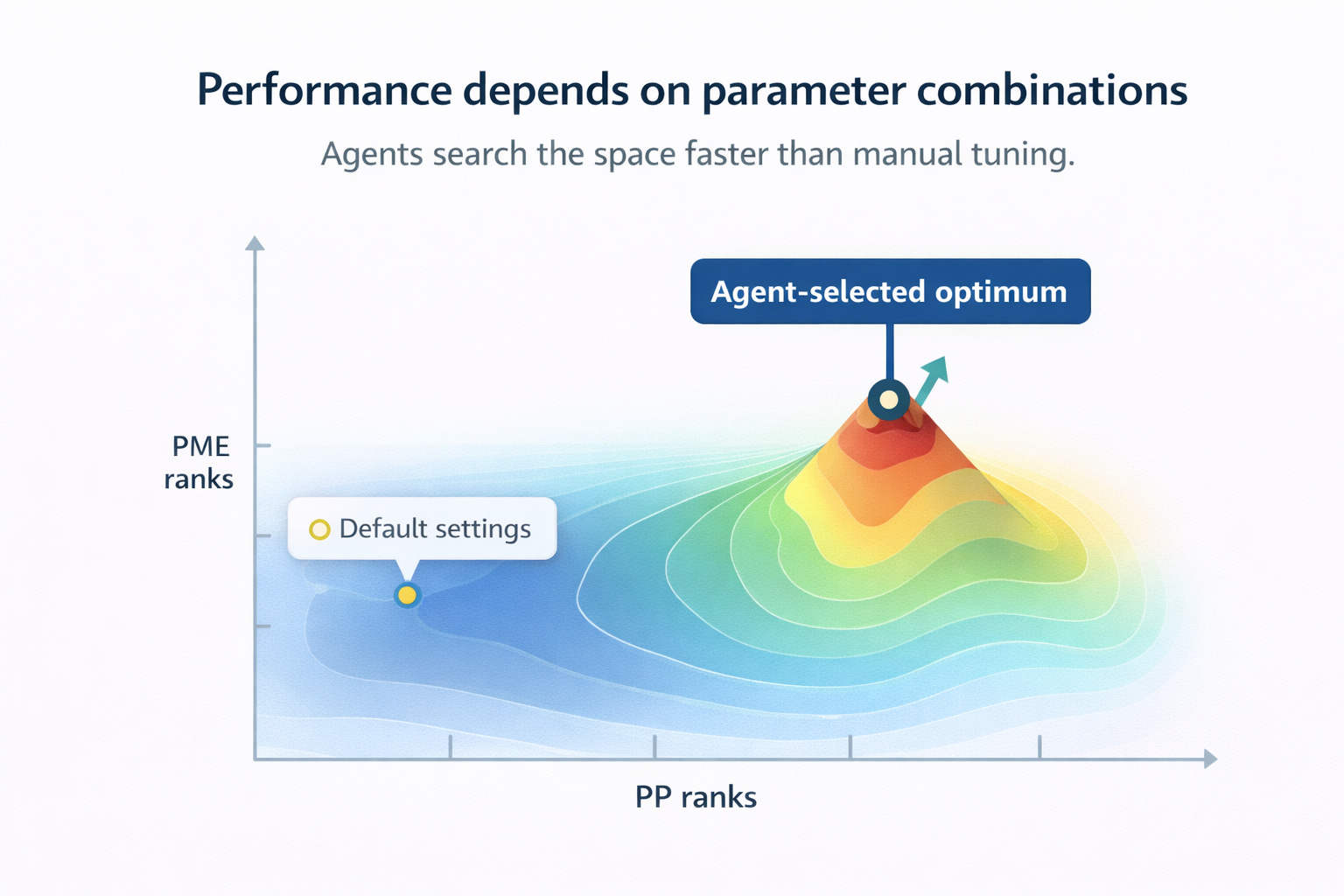
Figure: AI-driven parameter optimization outperforms default GROMACS settings by searching the full PP/PME configuration space.
Sivakumar et al. ‘s 2024 Artificial Intelligence Review shows ML models trained on MD datasets can predict optimal simulation parameters before runtime [6], shifting from reactive troubleshooting to proactive configuration maximizing accuracy and efficiency.
Automation That Understands the Science
Fully autonomous MD simulations are emerging. TorchMD (Thölke & De Fabritiis, 2022) and OpenMM 8 (Eastman et al., 2024) integrate ML potentials directly into simulation engines, enabling end-to-end differentiable workflows where force fields optimize during runtime [7][8]. Chaudhary et al.’s 2024 Current Opinion in Structural Biology review argues AI and quantum computing integration is “catalyzing a revolution in computational biology” [9].
Platforms like Pauling.AI orchestrate complete GROMACS pipelines complex selection, system preparation, energy minimization, equilibration, production, analysis via command interfaces. Manual gmx command chains now execute through parameterized workflows handling force field selection, water configuration, ion concentration, and simulation duration automatically. Multi-day workflows compress into hours.
The bottleneck shifts from technical expertise to scientific question formulation. Smaller labs without computational specialists gain access to workflows previously reserved for expert practitioners. GROMACS pipelines requiring weeks of manual iteration become accessible through automated systems designed for reliability and reproducibility at scale.
Intelligent automation is here. The question: adoption speed.
Stay Ahead of the Automation Wave
Manual MD is giving way to AI-driven workflows fast, right now. Subscribe for updates on computational-chemistry automation, intelligent simulation platforms, and the ways AI agents are reshaping molecular dynamics research.
References
[1] Gupta, A., et al. (2024). “ASGARD: An Automated System for GROMACS Analysis, Reporting, and Documentation.” Journal of Biomolecular Structure and Dynamics. https://www.tandfonline.com/doi/full/10.1080/07391102.2024.2439838
[2] Kutzner, C., et al. (2019). “More bang for your buck: Improved use of GPU nodes for GROMACS 2018.” Journal of Computational Chemistry. https://onlinelibrary.wiley.com/doi/full/10.1002/jcc.26011
[3] Lin, T., et al. (2025). “MDCrow: A Large Language Model-Based Agent for Molecular Dynamics Workflow Automation.” arXiv preprint. https://arxiv.org/abs/2502.00539
[4] Bignon, E., et al. (2023). “CHAPERONg: A tool to facilitate automation of GROMACS-based molecular dynamics simulations and trajectory analyses.” Computational and Structural Biotechnology Journal. https://www.sciencedirect.com/science/article/pii/S2001037023000466
[5] Abraham, M. J., et al. (2025). “GROMODEX: GPU-optimized molecular dynamics parameter exploration.” Journal of Chemical Information and Modeling. https://pubs.acs.org/doi/10.1021/acs.jcim.4c01423
[6] Sivakumar, S., et al. (2024). “Machine Learning in Molecular Dynamics Simulations: A Comprehensive Review.” Artificial Intelligence Review. https://link.springer.com/article/10.1007/s10462-024-10989-z
[7] Thölke, P., & De Fabritiis, G. (2022). “TorchMD: A deep learning framework for molecular simulations.” Journal of Chemical Theory and Computation. https://pubs.acs.org/doi/10.1021/acs.jctc.1c01126
[8] Eastman, P., et al. (2024). “OpenMM 8: Molecular Dynamics Simulation with Machine Learning Potentials.” Journal of Physical Chemistry B. https://pubs.acs.org/doi/10.1021/acs.jpcb.4c04662
[9] Chaudhary, N., et al. (2024). “Computational biology and chemistry: A tapestry of innovation.” Current Opinion in Structural Biology. https://www.sciencedirect.com/science/article/abs/pii/S0959440X24001258