
Running MD simulations with membrane proteins
By the Pauling.AI Team
Building a membrane system for molecular dynamics simulation often sounds simpler than it is.
In practice, there are many decisions that have to be made before the simulation can even begin. You need a lipid composition that makes sense for the target. The protein has to be oriented correctly inside the bilayer. The system needs to be equilibrated carefully, step by step, so it stays stable. Only after that can you start the MD simulation that gives you useful information.
That setup takes time. It also takes experience. And when something goes wrong early, you may not notice it until much later in the workflow.
This is especially important because many of the most relevant drug targets are membrane proteins. GPCRs alone represent roughly a third of all FDA-approved drug targets [1], and that does not include ion channels or transporters.
These proteins do not exist in isolation. They live inside membranes, and the membrane is not just a passive container.
The bilayer can apply lateral pressure on the protein. It can stabilize some conformations over others. It can also influence the shape and accessibility of the binding pocket in ways that are missing from a stripped-down crystal structure.
So, while simulating a membrane protein without its membrane can still provide useful information, it leaves out part of the biological context that may matter for drug discovery.
That is why we have been building membrane simulation support inside Pauling.AI Lab
Building the membrane around the protein..
The workflow starts with the protein target.
From there, Pauling.AI automatically builds the lipid bilayer around the protein, places the target in the correct membrane orientation, and parameterizes the full system for molecular dynamics.
Once the system is built, it goes through a staged equilibration that follows established GROMACS best practices, releasing restraints on the protein backbone, sidechains, and membrane lipids gradually so the membrane-protein complex settles into a stable, physically realistic state before the GPU-accelerated production run. The preparation matches the standard a computational chemist would apply by hand, so the results are suitable for downstream drug discovery work rather than just a visually plausible model, with the full equilibration protocol and simulation parameters available in our technical documentation.
The goal is not just to create a nice-looking membrane model. The goal is to make membrane-aware simulation practical inside a drug discovery workflow.
Because this is integrated into the existing Pauling.AI pipeline, the results connect directly with the same druggability analysis, scoring, structural review, and downstream prioritization used for other targets.
The biggest advantage is time.
Manual membrane setup can take days of careful work from a computational chemist. It requires knowledge of membrane systems, force fields, equilibration protocols, and common failure points. We have automated the parts that usually slow teams down, while keeping the physics and system preparation rigorous.
Starting with one of the hardest cases: GLP-1R
We used GLP-1R as one of our internal benchmarks.
GLP-1R is a class B GPCR. It has also become one of the most active areas in drug discovery, especially because of the interest in oral small-molecule agonists.
That makes it a strong test case for membrane simulation.
For GLP-1R, the binding site is not fully defined by the protein alone. The membrane environment helps shape how ligands approach, access, and interact with the receptor. This is exactly the kind of problem where a simulation without the membrane can give an incomplete view.
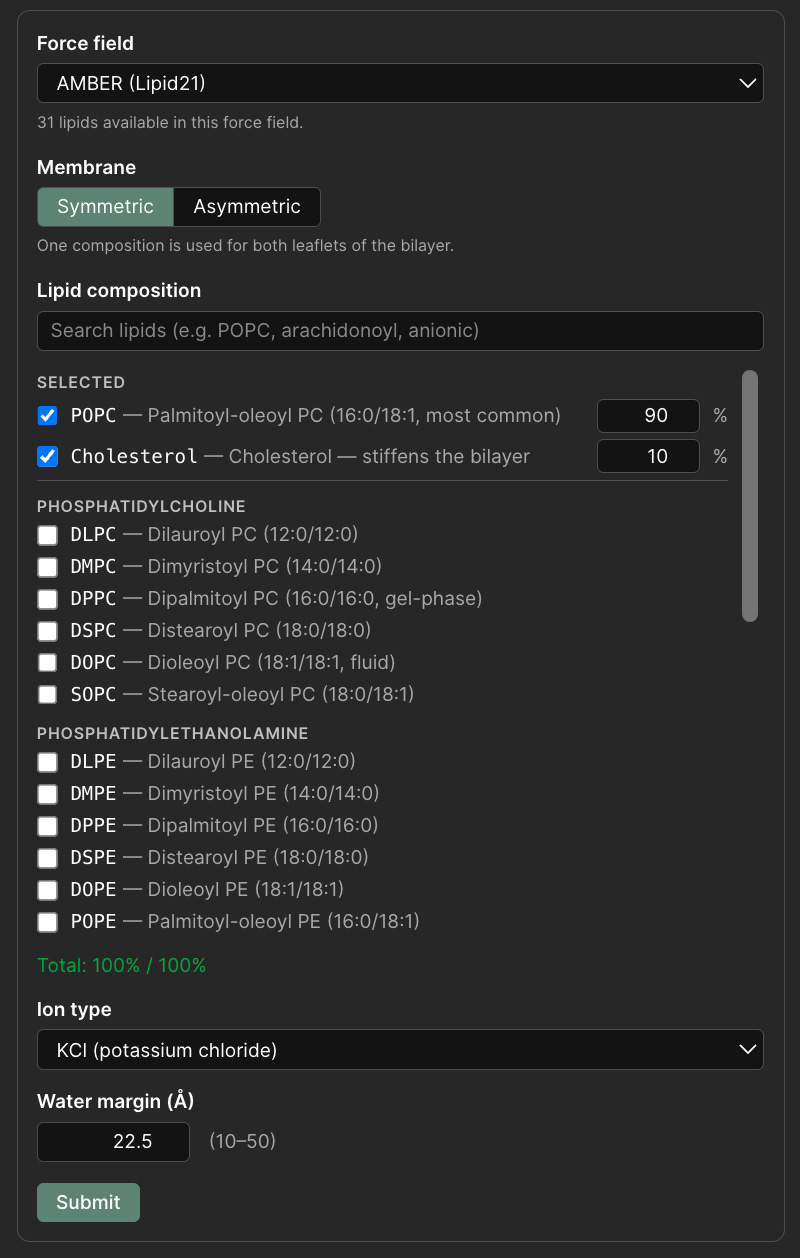
Membrane setup in Pauling.AI Lab: users can define the force field, symmetric or asymmetric bilayer composition, lipid ratios, ion type, and solvent margin before system parameterization.

GLP-1R (PDB: 9j1p) embedded in a lipid bilayer, The extracellular domain located above the membrane, the seven transmembrane helices span the bilayer, and the intracellular region extends below it.
The side view shows the overall architecture of the system: the receptor, the membrane, and the surrounding solvent.
The top-down view gives a different perspective. It shows the scale of the lipid environment around the receptor and makes clear that the protein is not being simulated in empty space, but inside the membrane context where its binding pocket actually exists.
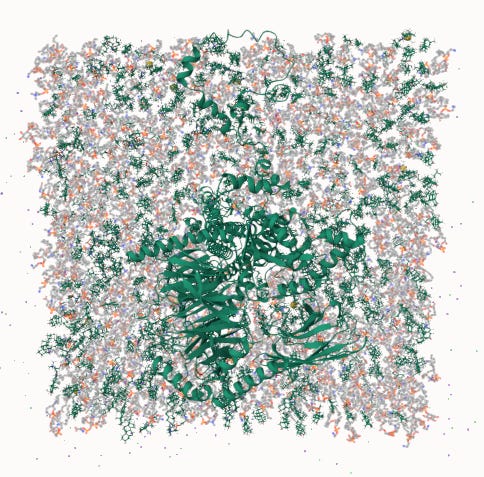
Top-down view through the membrane. The transmembrane domain of GLP-1R is shown at the center, surrounded by the full lipid bilayer. This is the environment the binding pocket experiences during simulation.
Who this is for
This workflow is designed for teams working on membrane protein targets, especially GPCRs, ion channels, and transporters.
For these programs, membrane simulation can help generate more meaningful structural and dynamic information earlier in discovery. Instead of waiting until a dedicated computational team can manually prepare a membrane system, the setup is handled directly inside the platform.
The result is MD-validated hit data in a more physiologically relevant context, earlier than membrane simulations would typically allow. The workflow applies to membrane proteins in general, with GPCRs, ion channels, and transporters being the most common target classes we support. For any of them, the main consideration is choosing an appropriate membrane composition, which the platform sets up automatically and which you can also define manually when a specific system calls for it.
If you want to see this workflow running on your target, reach out at contact@pauling.ai or visit pauling.ai.
Pauling.AI is a computational chemistry platform for drug discovery, offering an end-to-end pipeline spanning target modeling, pocket analysis, docking, molecular dynamics, and quantum mechanical scoring.
References
Sriram K, Insel PA. G Protein-Coupled Receptors as Targets for Approved Drugs: How Many Targets and How Many Drugs? Mol Pharmacol. 2018;93(4):251–258. https://doi.org/10.1124/mol.117.111062
GLP-1R structure (PDB: 9j1p). RCSB Protein Data Bank. https://www.rcsb.org/structure/9j1p