
From a UniProt ID to Ranked Hits: A Complete Walkthrough of Autochem's Docking Pipeline
By Pauling.AI Team.
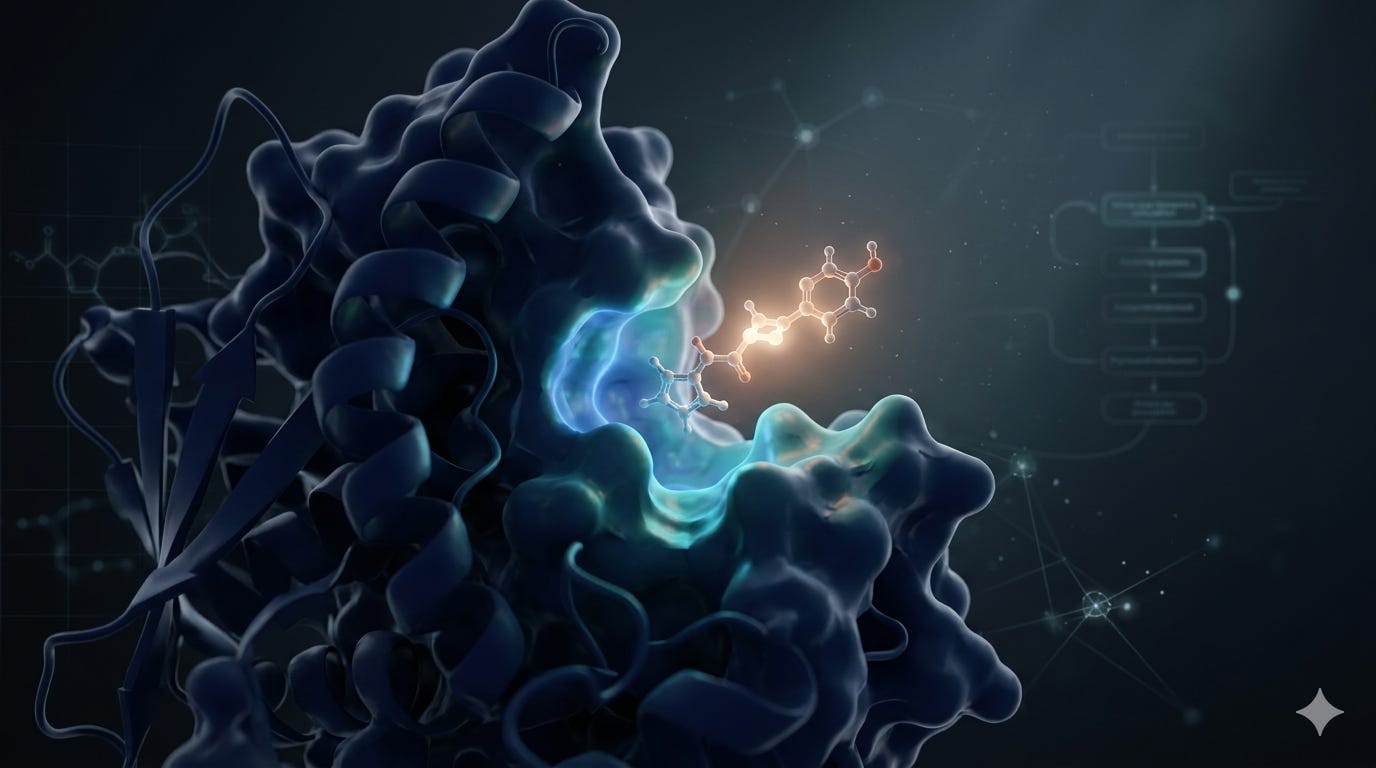
Structure-based virtual screening is only as reliable as the receptor model it runs on. A protein with missing hydrogens, strained geometry, or incorrect protonation at the active site will produce docking scores that don’t reflect real binding and the errors are rarely obvious in the output. Autochem’s docking pipeline handles the full sequence from target input to ranked, validated hits, with explicit quality control at each stage. This post describes what happens at each step and why.ure-based virtual screening is only as reliable as the receptor model it runs on. A protein with missing hydrogens, strained geometry, or incorrect protonation at the active site will produce docking scores that don’t reflect real binding and the errors are rarely obvious in the output. Autochem’s docking pipeline handles the full sequence from target input to ranked, validated hits, with explicit quality control at each stage. This post describes what happens at each step and why.
Preparation Determines Accuracy
Errors in receptor preparation propagate through every downstream calculation. A residue with the wrong protonation state shifts docking scores by several kcal/mol. A search box sized too conservatively excludes valid binding modes at the pocket periphery. A docked pose with an internal steric clash can receive a favorable score without being physically meaningful.
Autochem addresses this by treating preparation as the primary computational investment, with validation checkpoints before and after docking.
Step 1: Target Identification and Validation
Input can be a PDB ID, UniProt accession, UniProt entry name, or gene symbol. The identifier is validated against RCSB PDB, UniProt, and HGNC in priority order and normalized to a canonical type before any data is downloaded. Catching a mistyped or ambiguous identifier at this stage avoids wasted computation across all subsequent steps.
Step 2: Protein Metadata Download
The pipeline retrieves protein metadata from UniProt: gene names, organism, functional annotations, known isoforms, and database cross-references. This information supports chain selection downstream and flags targets with multiple orthologues or disease-associated variants that may require separate treatment.
Step 3: Protein Structure Acquisition
Experimental structures are downloaded from RCSB PDB in both mmCIF and PDB format (with Gemmi handling mmCIF conversion when needed). Multi-model structures such as NMR ensembles are split into individual conformations. The pipeline also queries OPM for membrane protein classification and NCBI for binding site annotations. When an experimental structure is available, it is always used X-ray and cryo-EM coordinates reflect the true atomic geometry of the binding site, including crystallographic waters, metal cofactors, and co-crystallized ligands.
Predicted structures from the AlphaFold Protein Structure Database are used when no experimental structure exists. Per-residue pLDDT scores identify regions of lower confidence typically flexible loops that may need additional scrutiny during pocket selection.
Step 4: Receptor Preparation: Fixing, Reducing, and Protonating

PDB files as deposited are generally unsuitable for docking. Three issues require systematic correction:
Missing hydrogens. Hydrogen atoms are not resolved at typical X-ray resolutions but are required by AutoDock-family scoring functions. Their absence also means hydrogen-bond geometry in the binding site cannot be evaluated correctly.
Missing atoms and residues. Disordered loops and terminal regions are frequently absent from deposited files. Gaps adjacent to the binding site create spurious openings that can accommodate poses the real protein would not permit.
Protonation states. Titratable residues histidine, aspartate, glutamate, lysine, arginine, cysteine, tyrosine adopt protonation states determined by local pH and electrostatic context. An incorrect assignment changes both the shape of the electrostatic surface and the hydrogen-bond donor/acceptor pattern of the active site, with direct consequences for scoring.
Four tools address these in sequence:
PDBFixer reconstructs missing heavy atoms from residue templates, fills terminal gaps, replaces non-standard residues, and strips crystallization artifacts.
Reduce (TRIM mode) places hydrogens using all-atom contact analysis. Histidine (protonatable at Nδ, Nε, or both) and the amide-bearing residues asparagine and glutamine — whose side chains can be flipped 180° without displacing any heavy atoms — receive particular attention.
PDB2PQR with the SWANSON force field assigns protonation states at pH 7.4 and adds complete hydrogen coverage. SWANSON is selected for its compatibility with AutoDock/Vina partial charges; substituting AMBER or CHARMM parameters introduces a systematic charge mismatch that degrades scoring in ways that are hard to trace.
OpenBabel converts PQR → PDB → PDBQT, AutoDock’s native format, which encodes torsional flexibility for each rotatable bond in the ligand.
Step 5: Receptor Energy Minimization
The operations in Step 4 residue replacement, atom addition, protonation reassignment perturb bond lengths, angles, and dihedral angles away from equilibrium values. Unrelaxed strain near the binding site introduces artificial steric penalties that affect which ligands score favorably.
Energy minimization with OpenBabel (MMFF94 force field) resolves this: steepest descent handles large gradients first, followed by conjugate gradient for convergence. Strain is reduced without displacing the structure significantly from its experimental coordinates.
Step 6: Structural Quality Control with MolProbity
The prepared receptor is validated with MolProbity, the tool used by the wwPDB for structural assessment of all deposited entries. The analysis covers four criteria: overall quality score as a PDB percentile, clashscore (steric overlaps per 1,000 atoms), Ramachandran backbone geometry, and rotamer quality for side chains. When multiple conformations have been generated, the pipeline selects the one with the best composite score.
Step 7: Binding Pocket Prediction with P2Rank
Binding site identification uses P2Rank (v2.5), a random forest model trained on structural and physicochemical features of protein surfaces derived from PDB examples of confirmed ligand binding. The model scores surface points by their likelihood of belonging to a druggable pocket, then clusters high-scoring regions into discrete predictions.
For each pocket, the pipeline defines a docking search box from the clustered surface points, expanded by 2 Å. The box size directly affects result quality: an undersized box clips valid poses; an oversized box dilutes the GPU search across empty space and raises the probability of sampling nonspecific sites.
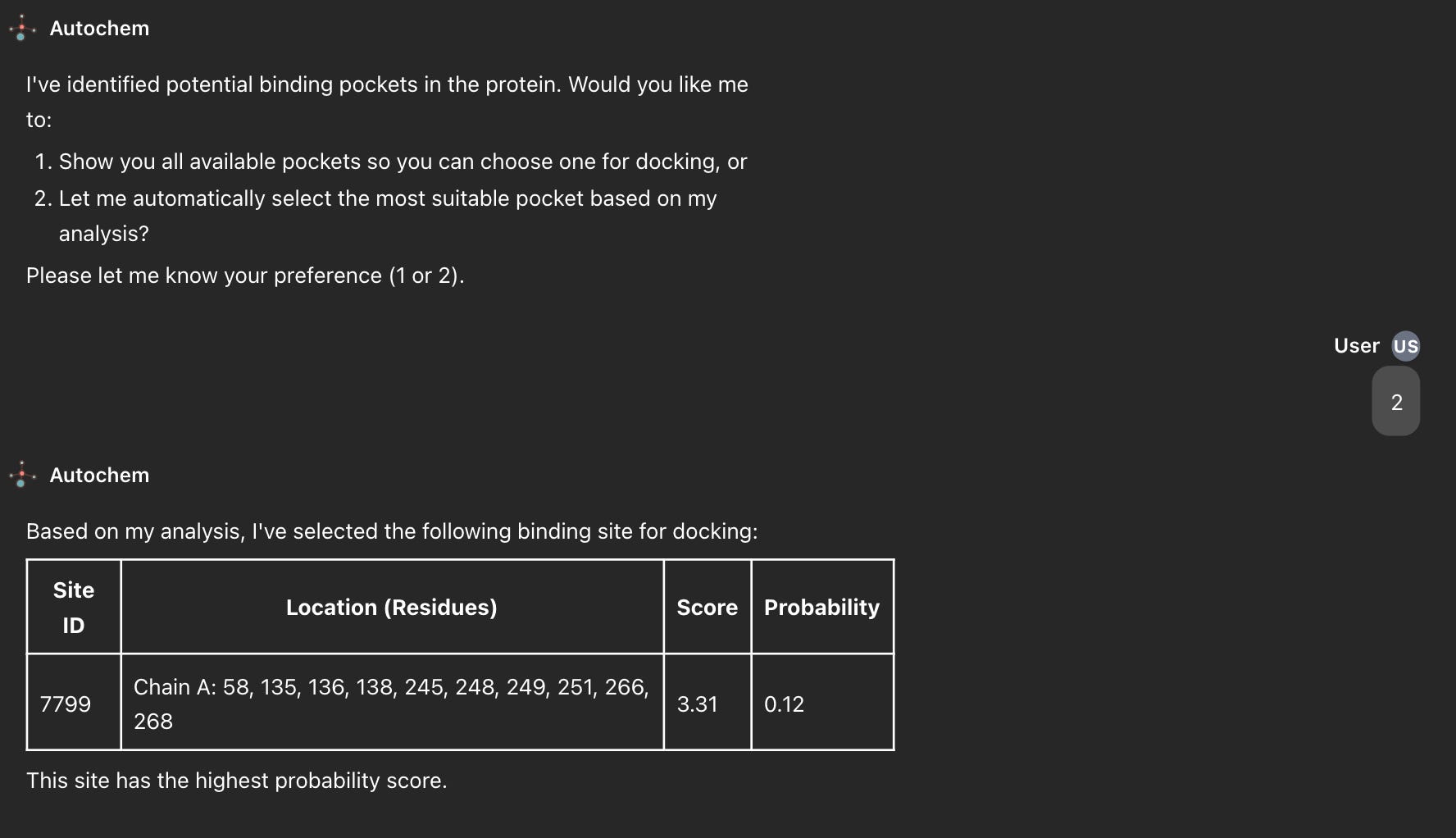
Step 8: Pocket Selection
Predicted pockets are presented as a 3D visualization overlaid on the receptor and as a ranked summary table. The user can select a pocket based on prior biological knowledge active site, allosteric site, secondary site or accept the top-ranked P2Rank prediction for automated workflows.
Step 9: Virtual Screening with UniDock
Virtual screening runs on UniDock, a GPU-accelerated implementation of the AutoDock Vina Monte Carlo conformational search. GPU execution enables batch docking of large libraries simultaneously, reducing runtime from days to hours relative to CPU-based Vina.
Before docking, the library is filtered to remove compounds with QED below 0.2, carbon-only nanostructures (which produce artifactually favorable scores due to their symmetric geometry), and endogenous metabolites.
Screening proceeds in two stages:
Stage 1 runs UniDock in fast mode across the full filtered library with a reduced per-ligand evaluation budget. Compounds scoring above −3.0 kcal/mol are removed. This stage reduces the library to a manageable subset for detailed evaluation.
Stage 2 re-docks the top 100 Stage 1 candidates with a substantially higher Monte Carlo budget and with explicit hydrogen atoms rather than the implicit representation used in Stage 1. Explicit hydrogens allow the scoring function to evaluate N–H and O–H bond directionality, improving accuracy for hydrogen-bond-dependent binding modes.
Step 10: Docking Quality Control with PoseBusters
Favorable docking scores are a necessary but insufficient criterion for hit selection. Scoring functions can reward poses that contain internal steric clashes, bond geometry violations, or atoms placed outside the binding site. PoseBusters applies physics-based checks to every pose: valence correctness, bond lengths and angles within expected ranges, no intraligand clashes, no clashes with protein heavy atoms, ligand center of mass within the declared pocket, and aromatic ring planarity. Poses failing any check are flagged for manual review before progressing to experimental follow-up.
Output
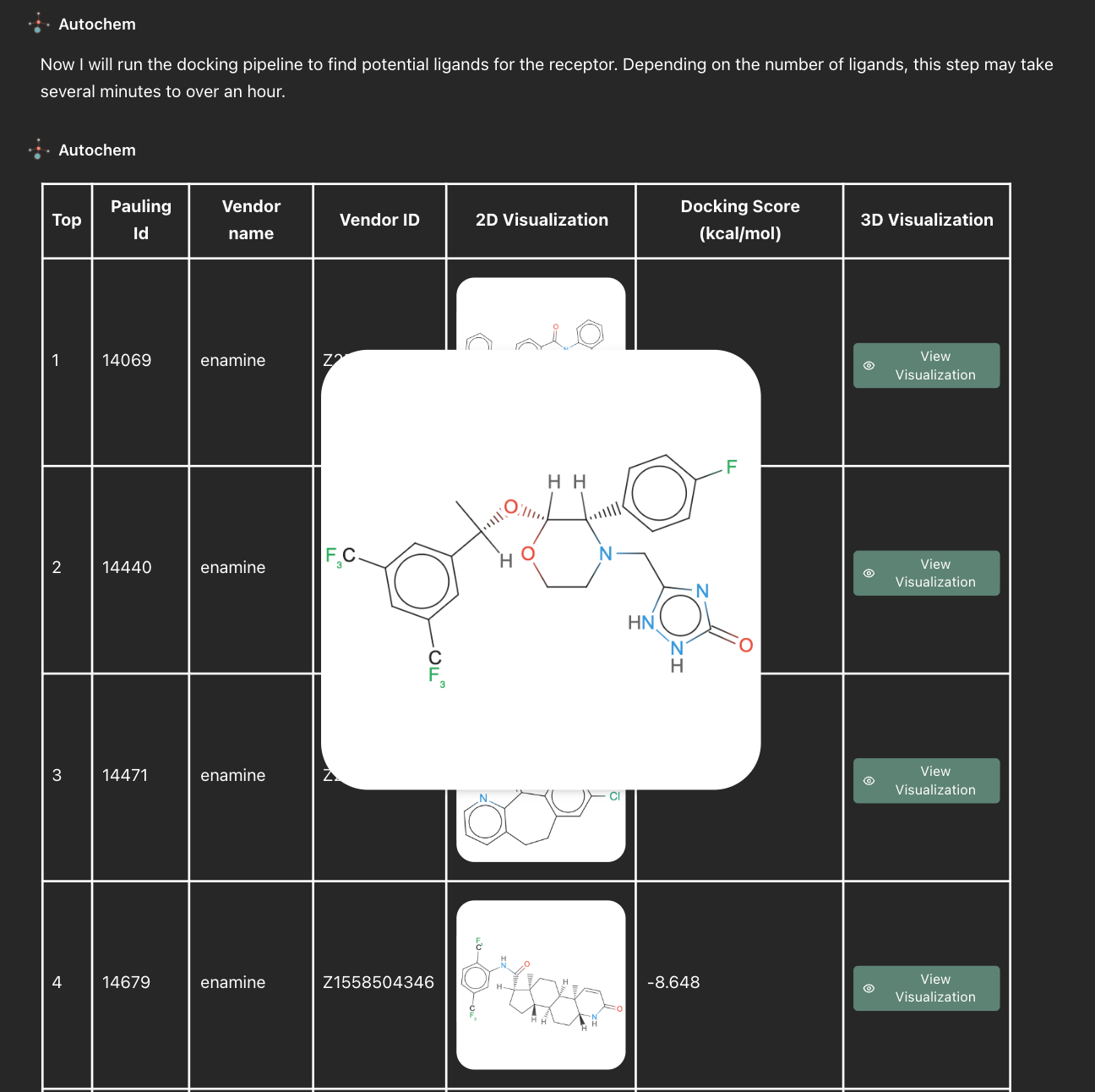
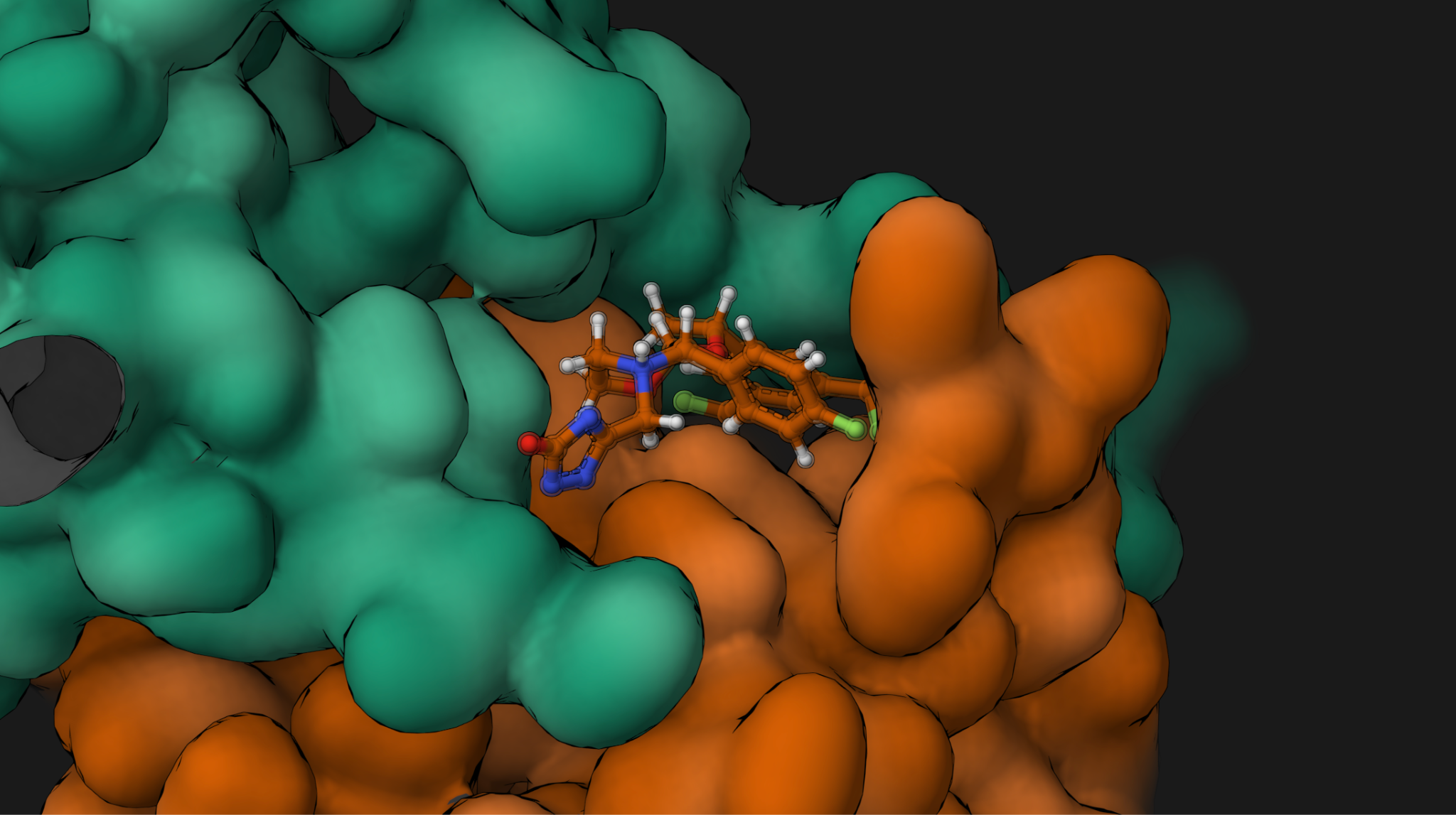
Autochem returns a ranked compound list with predicted binding affinities and per-pose validation status. Intermediate structures are stored at each step, and the full pipeline runs as distributed Apache Beam jobs on Google Cloud Dataflow with on-demand GPU provisioning for the docking stage. The workflow is reproducible and fully traceable from input identifier to final ranked hit.
Ready to run a virtual screen on your target? Start with Autochem →