
Designing for Selectivity: Counter-Screening Against JAK2 V617F
By Pauling.AI Team.
How Autochem’s selective counter-screening workflow helps researchers identify ligands that bind the mutant driver and leave the wild-type alone.
Finding a molecule that binds your target is one thing. Finding one that only binds your target, and not a dozen related proteins, is where things get interesting.
Counter-screening is how teams tackle that problem early: before a promising hit burns through expensive assays or makes it into an animal study.
The idea is straightforward, test your candidates against related proteins systematically, and see what you can rule out.
The execution is where it gets complicated.
We want to walk through a concrete example of how this works in Autochem, using a case that’s both biologically compelling and practically instructive: small molecules designed to hit mutant JAK2 V617F in polycythemia vera, while leaving wild-type JAK2 alone.
This post is a workflow demonstration, not a claim that docking alone can prove mutant selectivity. The goal is to show how Autochem’s counter-screening agent can help researchers generate and prioritize selectivity hypotheses early. Any promising docking result would still need to be confirmed with molecular dynamics and, ultimately, experimental wet-lab validation.
The biological story: why JAK2 V617F matters
Polycythemia vera (PV) is a myeloproliferative neoplasm a type of blood cancer and in the vast majority of patients, it’s driven by a single point mutation: valine to phenylalanine at position 617 of JAK2 (V617F).
That one swap locks the kinase in a constitutively active state, triggering uncontrolled red blood cell proliferation.
From a drug design perspective, this is actually a useful situation. The mutant protein is the driver. Wild-type JAK2 is doing normal, important things in healthy tissue.
So ideally, you want a molecule that preferentially engages V617F and mostly ignores the wild-type, reducing the risk of on-target toxicity where you don’t want it.
That kind of mutant selectivity is genuinely hard to achieve. What follows is a showcase of a workflow and a rigorous starting hypothesis not a claim that selectivity is easy or already solved.
Choosing the right pocket: JH2, not ATP
Most kinase inhibitors target the canonical ATP-binding pocket. For JAK2, that’s the JH1 kinase domain. But when the goal is to distinguish mutant from wild-type, the JH2 pseudokinase domain which is directly adjacent to V617F tells a far more interesting biological story.
Wild-type JAK2
Val617 in JH2 pseudokinase domain. Normal autoinhibitory interaction between JH1 and JH2. Kinase activity is tightly regulated.
Mutant JAK2 V617F
Phe617 disrupts the JH1–JH2 interface. Constitutive kinase activation. Altered pocket geometry in and around JH2.
That structural difference is exactly what you want to exploit. By targeting JH2 rather than the shared ATP site, the docking workflow has a structurally meaningful basis for distinguishing mutant from wild-type, not just comparing scores against an arbitrary reference.
The counter-screening workflow, step by step
Autochem runs two parallel docking and analysis pipelines one against mutant JAK2 V617F, one against wild-type JAK2 using the same set of candidate ligands.
Here’s how it unfolds:
01 - Target preparation
Both structures are loaded and processed. The JH2 pseudokinase pocket is defined as the binding site for both targets.
02 - Ligand library setup:
A set of candidate small molecules is queued, from an in-house library, a virtual screen hit list, or de novo generated structures
03 - Parallel docking runs
Each ligand is docked independently into both the mutant and wild-type pockets. Autochem handles both pipelines automatically, no manual hand-off between steps.
04 - Selectivity scoring:
Docking scores for each ligand are compared across the two targets. Molecules with a favorable score differential, stronger predicted binding to V617F, are flagged as selectivity candidates.
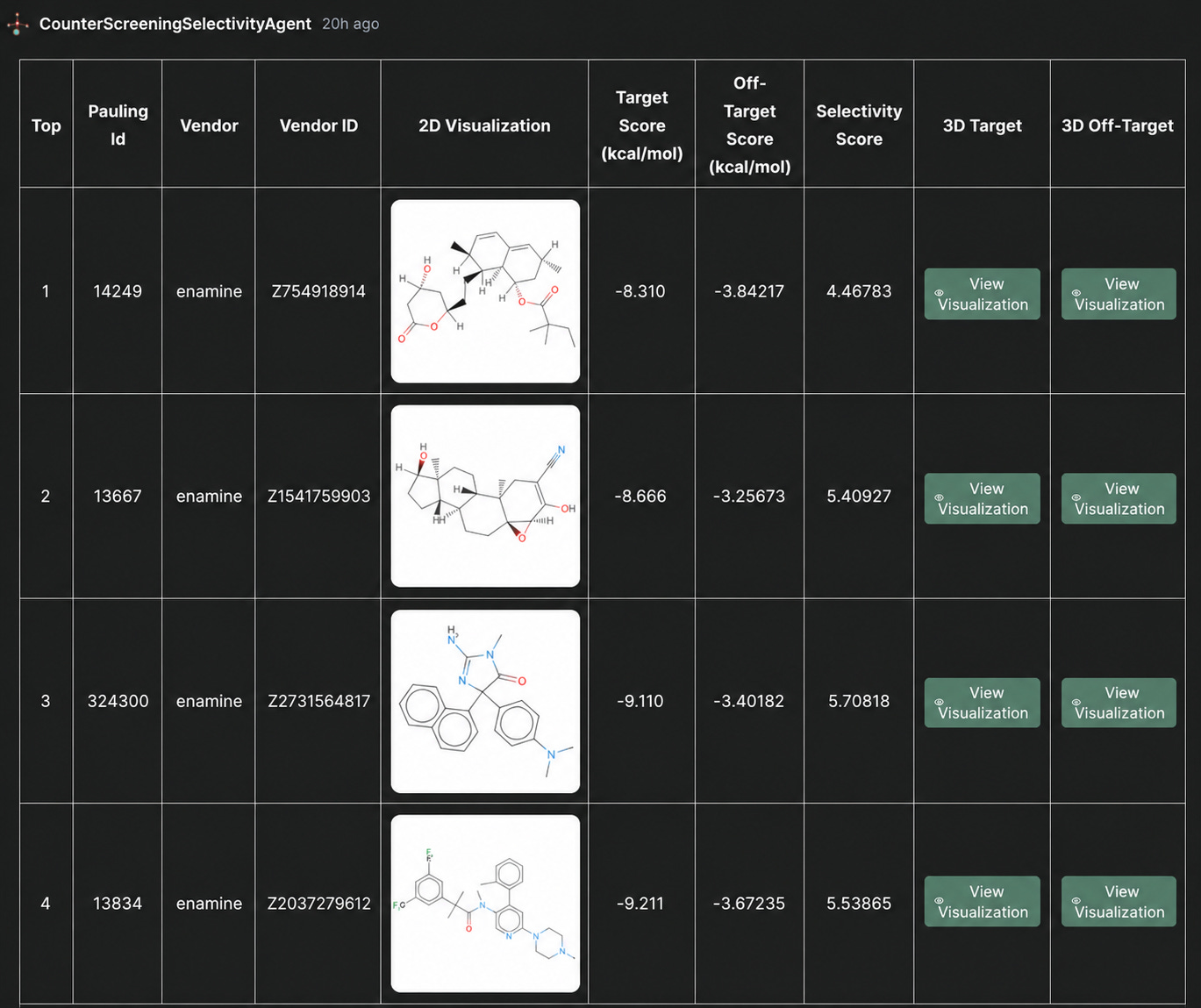
Figure 1. Top-ranked ligands from the counter-screening workflow, sorted by selectivity score and showing target, off-target, and selectivity scores.
05 - MD validation
Top candidates can be passed into short molecular dynamics simulations to test binding stability in each context, adding a dynamic dimension to the static docking picture.
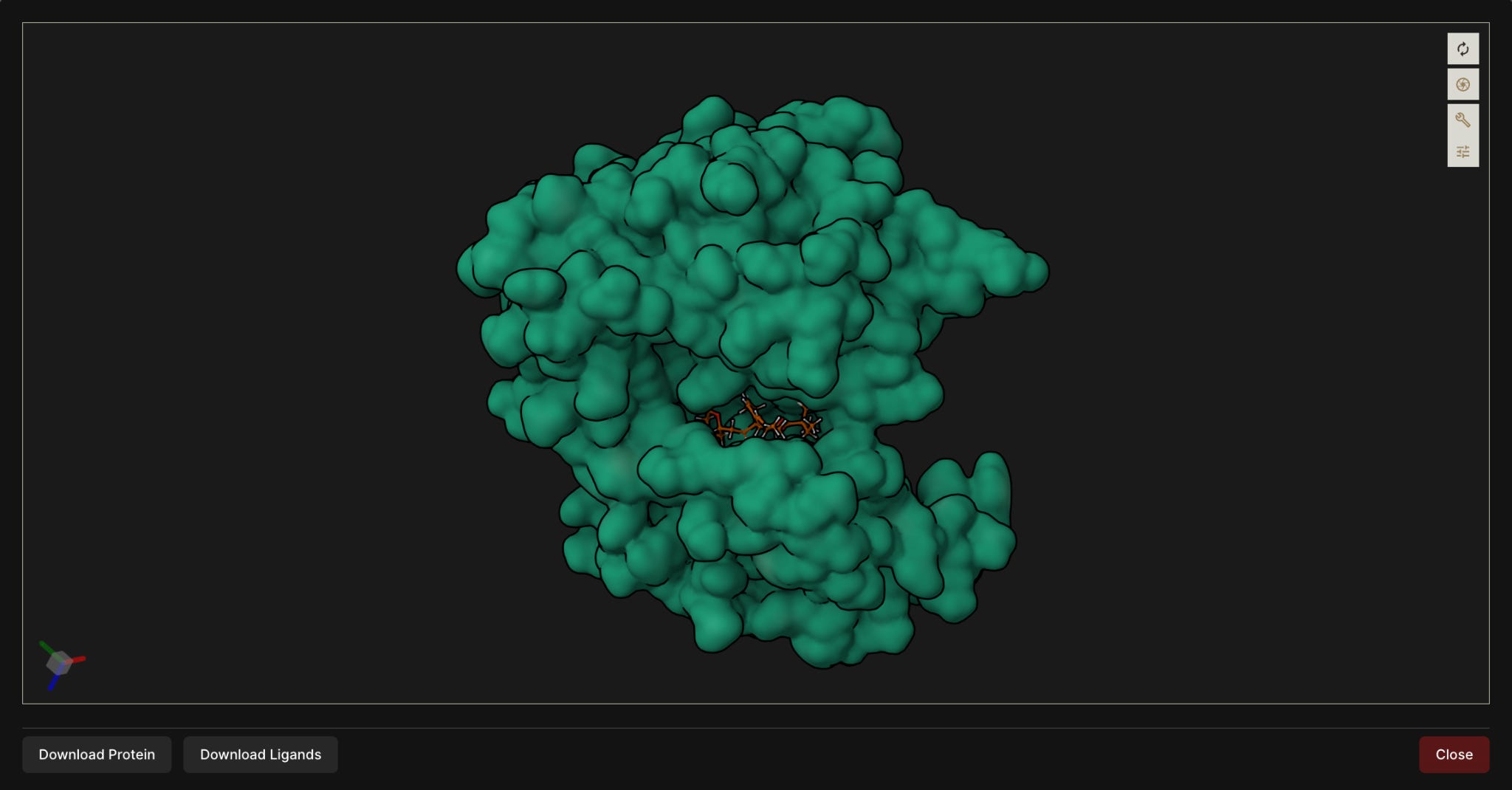
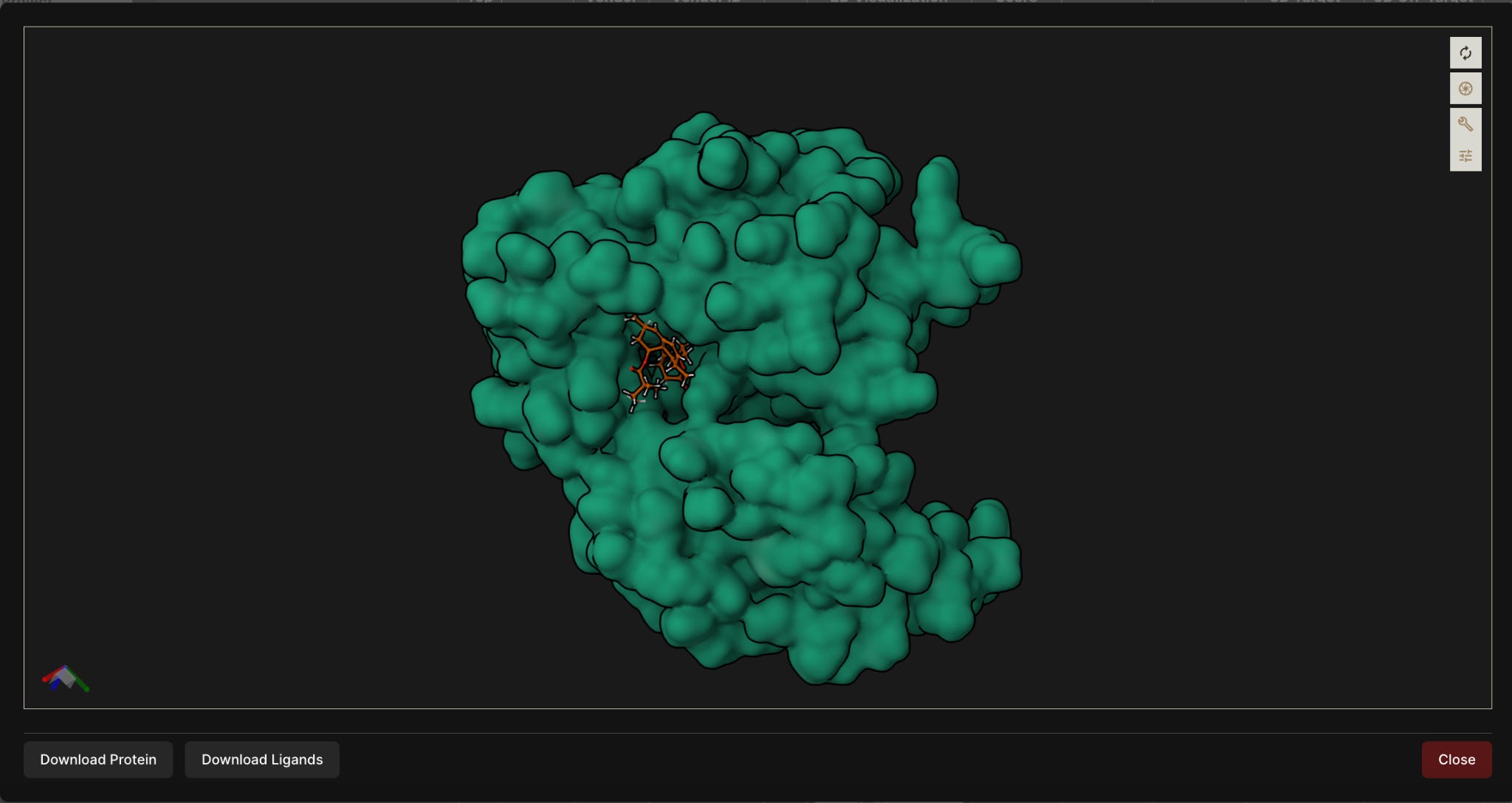
Figure 2. 3D visualization of a top-ranked ligand docked into the selected JH2 pocket of the desired JAK2 V617F target (above) and wild-type (below).
What the output looks like
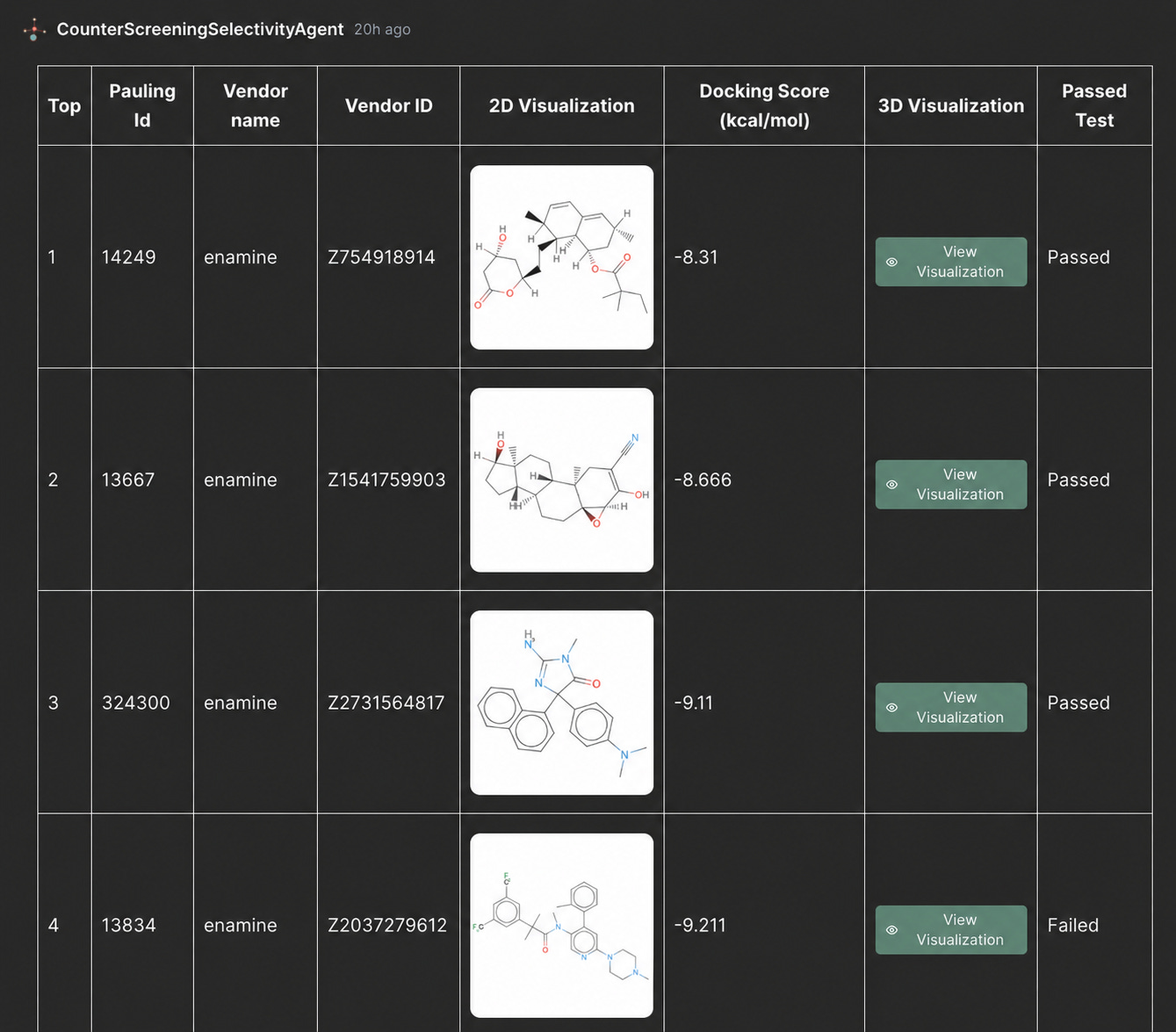
Figure 3. PoseBusters quality-control output for selected top-ranked ligands, showing whether the docked poses pass pose-quality checks before MD and wet-lab validation.
Before advancing selected hits into molecular dynamics or experimental follow-up, Autochem can add a pose quality-control step.
Here, the top-ranked ligands from the counter-screening output are evaluated with PoseBusters to check whether the predicted docking poses are geometrically and chemically reasonable. This helps ensure that candidates are not prioritized based on docking scores alone, but also pass an additional pose-quality filter before deeper validation.
The workflow returns a ranked comparison: for each ligand, a score against mutant JAK2 V617F a score against wild-type JAK2, and the selectivity ratio, the difference between the two scores.
That’s your starting point for a medicinal chemistry conversation. A large score differential doesn’t guarantee selectivity in a cell. But it gives your team a prioritized, computationally grounded hypothesis to test which is exactly what you want at this stage.
Why this matters beyond polycythemia vera
Mutant-selective targeting keeps coming up across oncology. EGFR T790M, KRAS G12C, IDH1 R132H the list of clinically relevant driver mutations is long, and the bottleneck is often not biology but tooling: how quickly can a team generate meaningful selectivity hypotheses early in discovery?
The counter-screening workflow shown here generalizes naturally. Swap in any pair of related targets mutant vs. wild-type, paralog vs. paralog, pathogen vs. host ortholog and the same orchestrated pipeline applies. The goal is always the same: find the molecules with the best differential signal, as early as possible.
Autochem’s counter-screening workflow is part of the broader Autochem platform, which orchestrates 150+ computational tools across target modeling, virtual screening (docking), molecular dynamics, and analysis pipelines.
Interested in running a selectivity counter-screen for your target? Get in touch with the Pauling.AI team.
JAK2 inhibitorspolycythemia veracounter-screeningmutant selectivitymolecular dockingAutochemdrug discovery workflow