
A multi-stage docking workflow, and why regular docking needs structure
By the Pauling.AI Team
Docking is often described as if it were a single step.
Take protein. Take a molecule. Run docking. Read the score.
But in practice, useful docking depends on a chain of decisions that happen before and after the docking calculation itself. The receptor needs to be prepared. The binding site needs to be selected. Candidate molecules need to be ranked, checked, and inspected in 3D.
A docking score alone is not enough if the protein structure was not prepared correctly, the search space was poorly defined, or the final pose is not structurally reliable.
That is why Autochem treats regular docking as a multi-stage workflow rather than a one-step calculation.
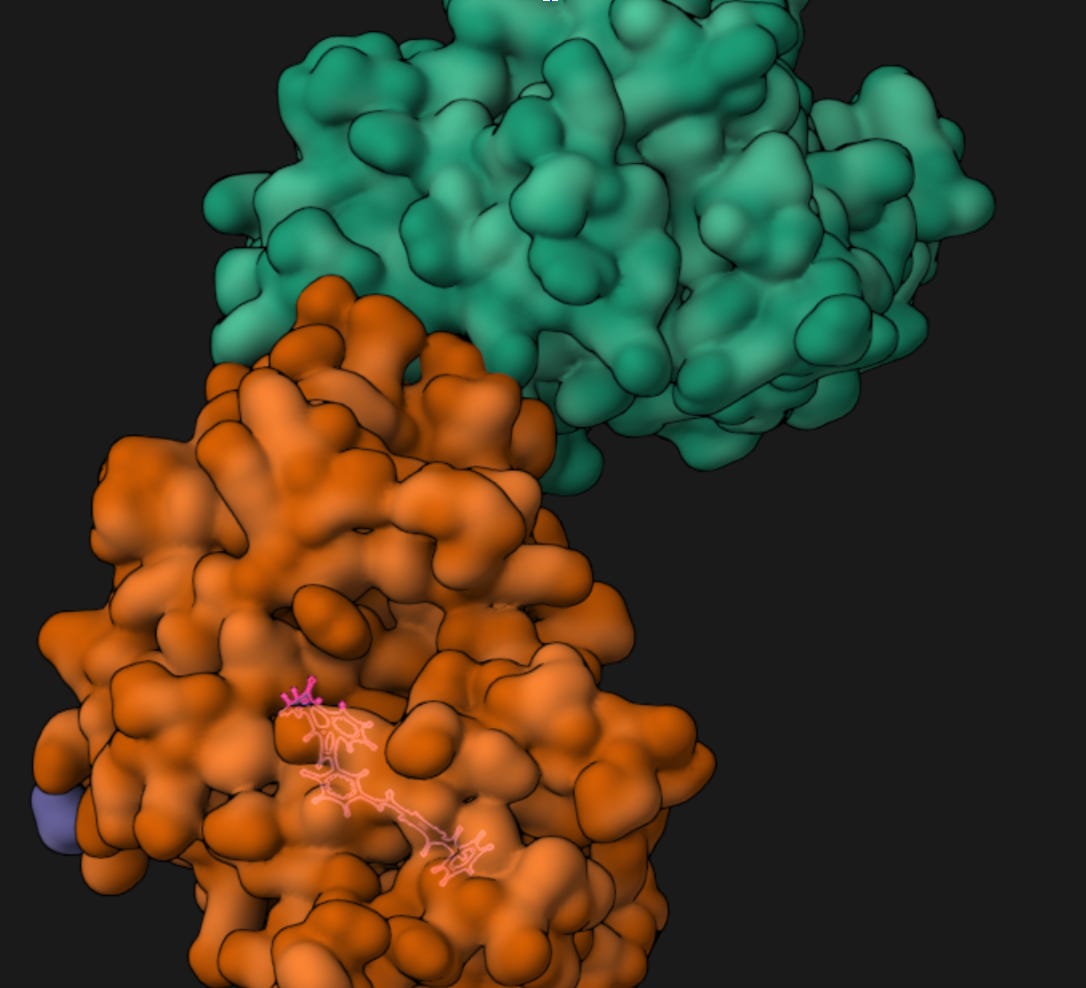
Autochem visualizing the protein structure before docking.
Why we chose c-Abl as the test target
For this workflow, we used the c-Abl tyrosine kinase domain, starting from PDB structure 1IEP.
c-Abl is a strong target for demonstrating a regular docking workflow because it is a well-studied kinase with a clear small-molecule binding context. The structure is associated with STI-571, also known as imatinib, one of the best-known examples of structure-guided kinase inhibition.
That makes c-Abl a useful system for showing how a docking workflow moves from a known protein structure into a practical computational process.
The goal here is not to claim a full benchmark or a complete redocking validation. Instead, the goal is to show the stages that make regular docking more interpretable: target validation, receptor preparation, binding-site selection, docking, ranking, pose-quality control, and visual inspection.
Stage 1: target validation and receptor preparation
The workflow begins by validating the target.
Autochem starts from the PDB identifier and confirms that the structure is valid before downloading the protein data. From there, it prepares the receptor for docking by assigning a physiological pH, running protonation steps, and minimizing the receptor structure to address remaining geometry issues.
This matters because raw protein structures are not always docking-ready.
Missing hydrogens, unclear protonation states, and local geometry problems can all affect how a ligand is evaluated inside the pocket. If those issues are ignored, the docking result may look precise while being built on an unreliable input.
Before any score is produced, the receptor has to become a chemically usable structure.
Fig 1: Autochem validates the target structure and prepares the receptor through pH assignment, protonation, and energy minimization.
Stage 2: binding-site selection
Once the receptor is prepared, the workflow needs to define where docking should happen.
Autochem identifies a candidate binding site and reports the residues that make up the selected region, along with a site score and probability. This step narrows the search to a structurally plausible region of the protein.
That matters because docking is only as useful as the search space it explores.
If the binding region is too broad, the calculation may waste time exploring irrelevant regions. If it is too narrow or poorly placed, the workflow may miss useful poses. Binding-site selection helps focus the docking calculation before molecules are evaluated.

Fig 2: Autochem selects a high-probability druggable binding site and identifies the list of residues that define the docking region.
The goal at this stage is not to choose the final molecule. The goal is to define the right structural context for the search.
Stage 3: docking and ranked candidate generation
With the receptor prepared and the binding site selected, Autochem runs the docking pipeline.
The output is a ranked list of candidate molecules. Each result includes molecular identifiers, a 2D structure, a docking score, and the option to inspect the predicted 3D binding pose.
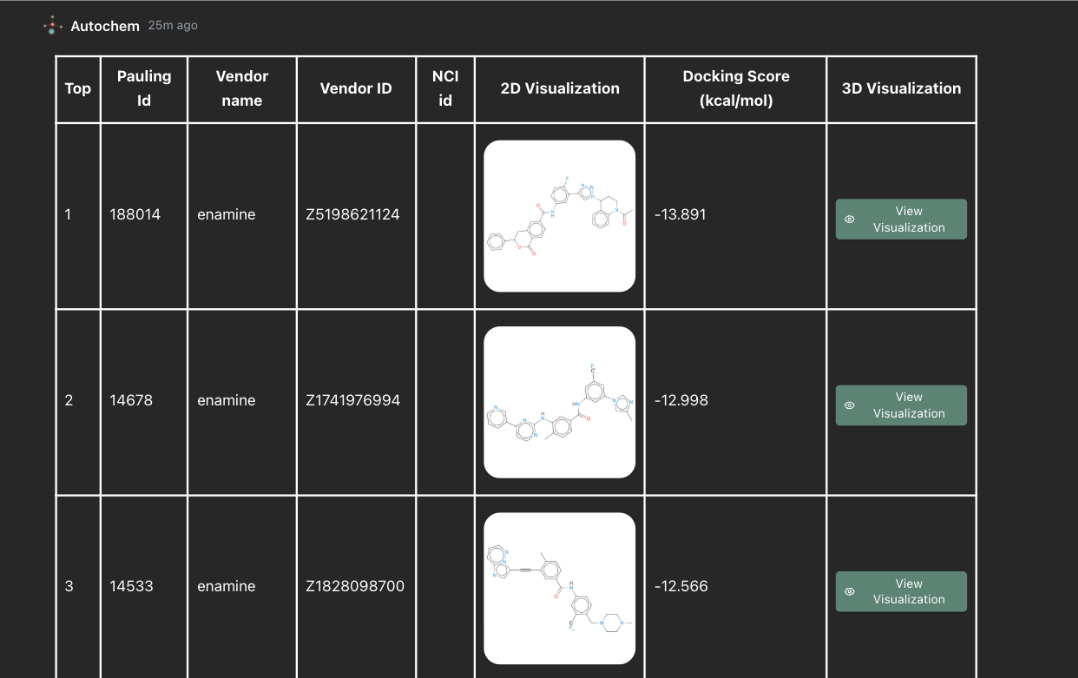
Fig 3: Autochem ranks the top docking hits by score and links each compound to its 2D structure and 3D pose visualization, allowing users to move from numerical ranking to structural inspection.
This ranking is useful, but it is not the final answer.
Docking scores help prioritize candidates, but they should not be treated as absolute truth. A strong score does not automatically mean a molecule is chemically convincing inside the binding site. It simply means the candidate is worth looking at more carefully.
That is why the workflow continues beyond ranking.
Stage 4: pose-quality control
After docking, Autochem runs quality-control checks on the predicted poses.
This step helps assess whether the docking outputs are structurally reliable enough for further review. Instead of asking the user to trust a score alone, the workflow checks whether candidate poses pass basic reliability filters before deeper inspection.
This matters because docking can produce attractive-looking scores for poses that are not chemically or geometrically convincing.
A molecule may score well while adopting a strained pose, forming unrealistic contacts, or fitting the pocket in a way that does not make structural sense. Pose-quality control helps reduce that risk before candidates are carried forward.
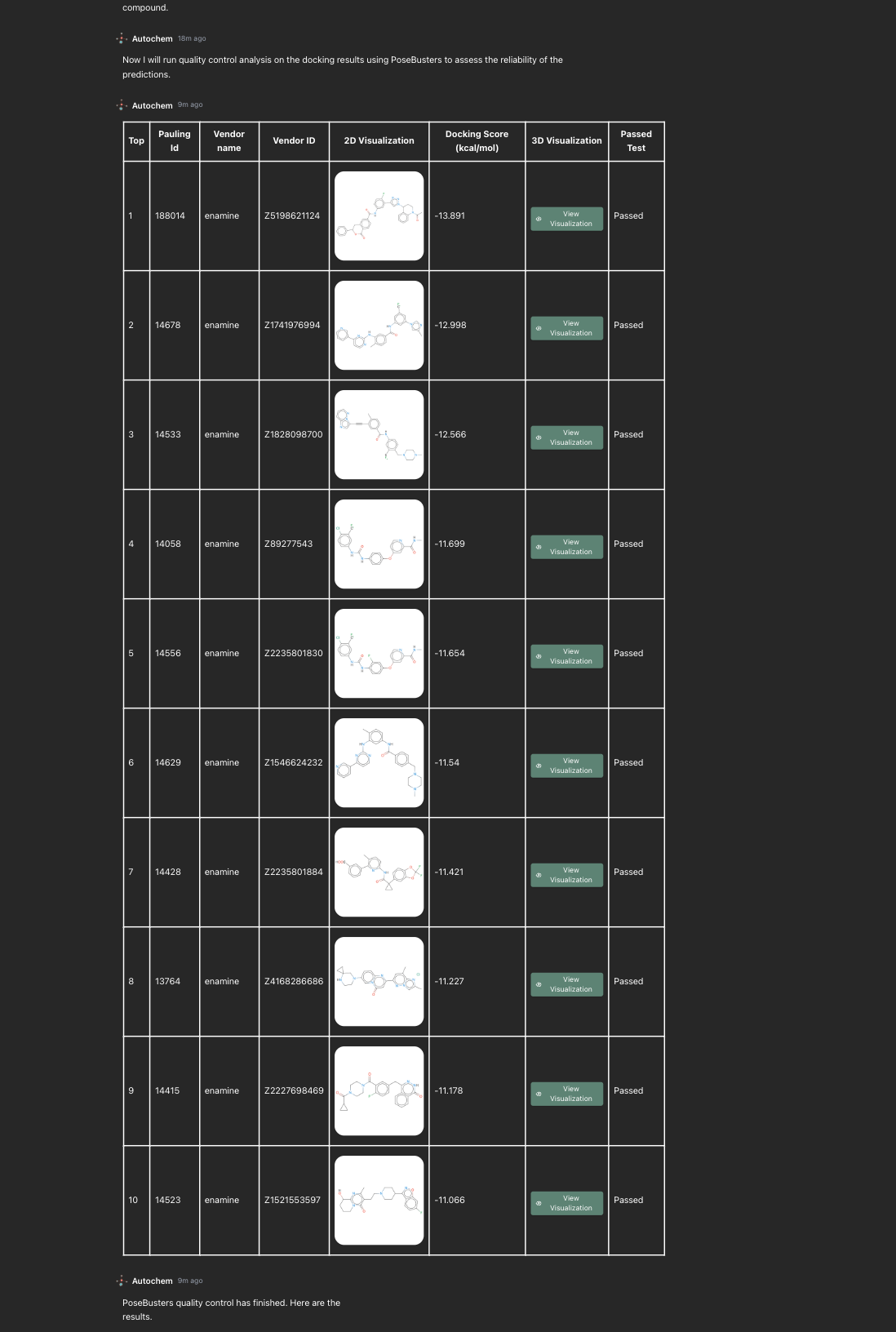
Fig 4: The final table of docking run produces a ranked shortlist of candidate molecules with 2D structures, docking scores, links to 3D inspection and quality control based on Posebusters validation.
Stage 5: visual inspection
The final step is visual inspection.
Autochem allows candidate molecules to be reviewed in 3D inside the binding site. This is where the ranked output becomes chemically interpretable.
A docking result should answer more than one question.
It should not only ask, “What scored best?”
It should also ask, “Does the pose make sense?”
Does the molecule occupy the intended pocket?
Is the binding mode plausible?
Is this candidate worth taking into a more detailed computational or experimental follow-up?
Visual inspection brings the workflow back to structure.
Fig 5: 3D inspection helps connect docking scores back to the physical structure of the protein and binding site.
What the workflow shows
The value of a multi-stage docking workflow is not that any single step is perfect.
Its value is that each stage reduces a different kind of uncertainty.
Target validation reduces input risk.
Receptor preparation reduces chemistry and geometry risk.
Binding-site selection reduces search-space risk.Docking generates a ranked shortlist.
Pose-quality control reduces false confidence.
3D inspection brings the result back to structure.Together, these stages make regular docking more practical and more interpretable.
The point is not just to produce a docking score. The point is to produce a result that a scientist can inspect, question, and prioritize.
That is the argument for a multi-stage docking workflow.
Not docking as a black box.
Not docking as a single score.
But docking is a structured process, where each stage makes the next one more useful.